


一、全新检查流程引发检查依据的迭代!
· 旧:检查完全基于21 CFR Part 820即QSR,包括多个FDA专有术语与要求。
· 新:QMSR自身并不涉及具体条款,而是直接引用纳入ISO 13485:2016的体系要求,并且叠加FDA于QMSR保留的特定法规要求(例:记录保存语言、某些定义、部分Part 803/806报告义务等)。
· QMSR时代下FDA检查将根据“ISO 13485:2016条款 + FDA附加要求”实施。
· FDA原先使用多年的《质量体系检查技术》(QSIT) 方法正式退出历史舞台,崭新的《医疗器械制造商检查合规程序手册》(7382.850)闪亮登场!
为聚焦风险并反映质量管理体系各流程之间的关系,《以风险为基础的检查流程》(如下图所示)将QMSR要求归纳为6大质量管理体系领域、4项其他适用的FDA要求:
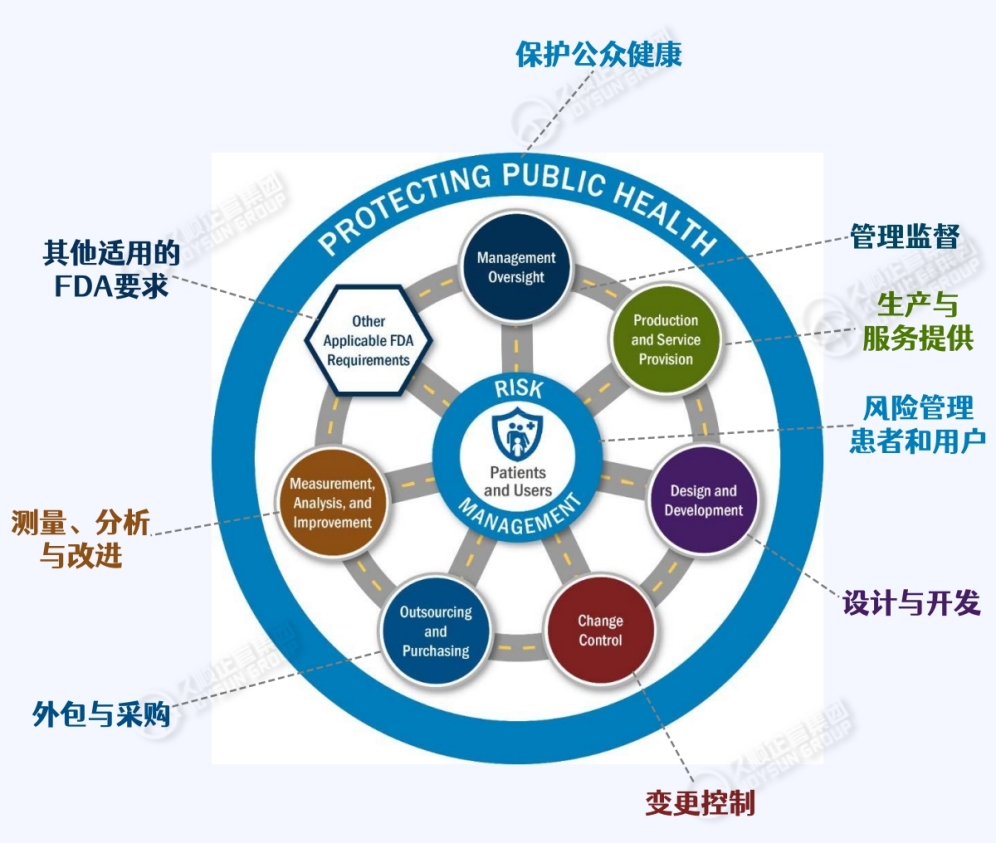
·6大领域分别为:管理监督、生产与服务提供·、设计与开发、变更控制、外包与采购、测量/分析与改进。
·4项要求分别为:其他适用的FDA相关要求(医疗器械报告\更正与移除报告\医疗器械追踪\医疗器械唯一标识)。
·核心为:以患者和用户为中心的风险管理。
·目标与作用为:保护公众健康。
二、检查方法的调整
· FDA检查人员将接受ISO 13485标准及体系法规新旧版区别的培训。
· FDA已更新其检查指南与工具(如上所述),即已修改QSIT方法,适应ISO 13485结构。
· QMSR下的FDA现场检查可能使用以ISO 13485条款为基础的检查清单,并重点关注FDA特意保留或新增要求。
三、全新的检查重点
1. 整合风险管理
ISO 13485明确要求在设计开发、生产、采购等全过程贯穿实施风险管理,并密切关联ISO 14971风险管理标准,FDA检查趋向更系统化、结构化,不再停留于设计阶段。
2.客户抱怨及反馈
· ISO 13485有着更宽泛的“反馈” 范围要求,涉及:主动收集信息(调查、调研),而QSR820旧规强调“抱怨的处理”。
· FDA将检查企业是否建立系统性的反馈机制,并确保其信息被输入至改进过程与风险监测。
3.验证与确认的变化
· 部分术语和范围发生变化(软件验证、灭菌过程确认等)。
· QMSR新规下的检查将根据ISO 13485语境结合FDA期望实施检查。
4. 监管报告要求的保留
· 需注意FDA特有要求依旧保留:MDR不良事件报告、召回报告、510(k)/PMA相关报告义务,依旧在QMSR框架下得到满足。
· 检查时需特别注意上述法规报告流程是否已被整合至体系中,且记录保存符合FDA期限要求(区别与ISO要求)。
5. 管理责任与质量方针
· ISO 13485强调建立质量目标、确保资源、推动改进中管理层的责任。
· 检查中可能更加重视管理评审输入输出的完整及有效性。
四、文件和记录要求
· 根据ISO 13485 4.2.5确定保存期限并符合适用的法规要求,同时需满足820.35对特定记录内容的补充要求。
· 语言要求:FDA要求有关记录能供其审核,必须被准确翻译为英文(通常要求关键记录有英文版本或可快速提供译文)。
· 文档结构:企业可能已根据ISO 13485建立手册与程序,但应证明其内容也满足FDA特定条款(可通过对照表或补充程序实现)。
五、QMSR下现场检查的企业应对方案
1.体系转换:QMSR与ISO 13485对照差距分析,将现有QSR体系更新为融入ISO 13485并符合FDA特定要求。
2.培训:使得制造商内部相关人员(特别是管理层、QA、RA)领会新旧法规差异和FDA保留的要求。
3.双语言文档:重要的质量记录与体系文件应当便于FDA检查(英文或可快速翻译)。
4.模拟检查:依据ISO 13485+FDA特定要求,邀请第三方法规辅导机构开展内部审核或模拟FDA检查。
↗FDA体系升级,就选久顺企管!30年老牌体系辅导专家,为您提供高效率定制级QMSR体系过渡服务,包括:法规差距识别及分析/提供优化升级方案/法规培训/英语文件编制/现场英语陪同迎审/全过程合规策略支持与辅导等。
>久顺是值得信赖的体系助手! 可提供:建立并维护国内质量管理体系/ISO13485体系/美国QSR820体系/MDSAP/质量体系培训&咨询&辅导等。
