


久顺经常收到这样的问询:MDR法规下如何准确选择医疗器械符合性评估路径?
首先,应明确器械在MDR下的分类。按照REGULATION(EU) 2017/745附录8,根据器械预期用途和固有风险,医疗器械大体被分为I、IIa、IIb 和III类;相同的器械由于不同的分类标准可能会被分到不同的类别,应选择最高等级的分类。
随后,以器械分类为基础,选择对应的符合性评估路径。
注意:同一类器械可能不止一种合法符合性评估路径,此时需要根据器械特点及制造商自身情况,选择更合适、更方便、更经济的符合性评估路径。针对于此,本期按照不同类别器械分享相应选择技巧。
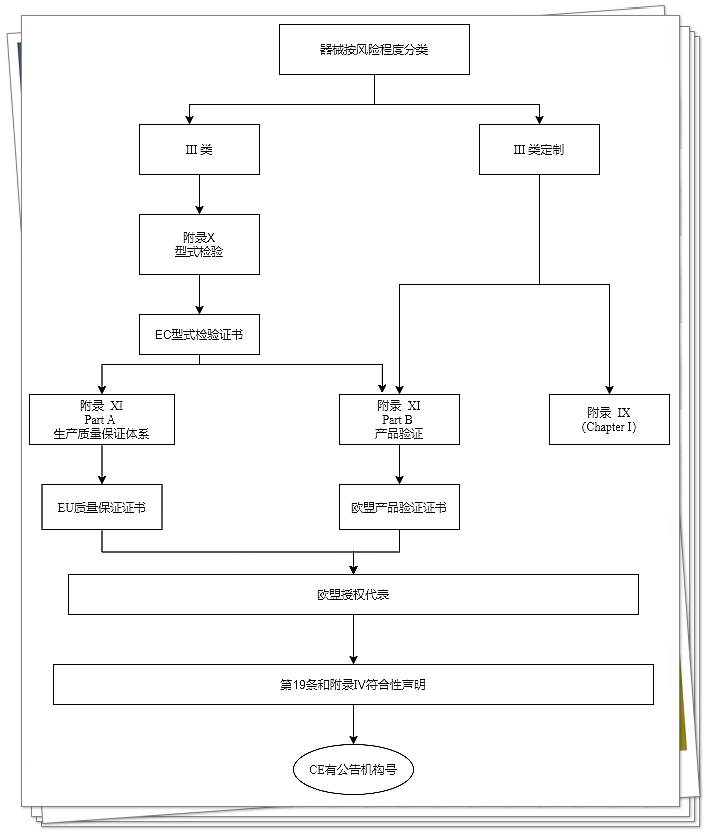
III类器械
III类器械(非客户定制类或研究类器械)的制造商,如果其质量体系较完善,可根据MDR附录9规定进行符合性评估。按此符合性评估路径,制造商需建立、记录和实施一套合格的质量管理体系,并保持其在相关器械整个生命周期内有效运行。
如果器械单件价值较高,生产周期较长,产量又极低,选择附录10和附录11的符合性评估路径则更合适。
欧盟型式试验是指欧盟公告机构确认器械性能并颁发合格证书的过程,包括:技术文档及符合法规规定的器械代表性样品。
基于产品合规性验证的符合性评估,目的是确保器械符合已发布的EC型式检验证书中所说明的形式,并满足法规中适用的规定要求。
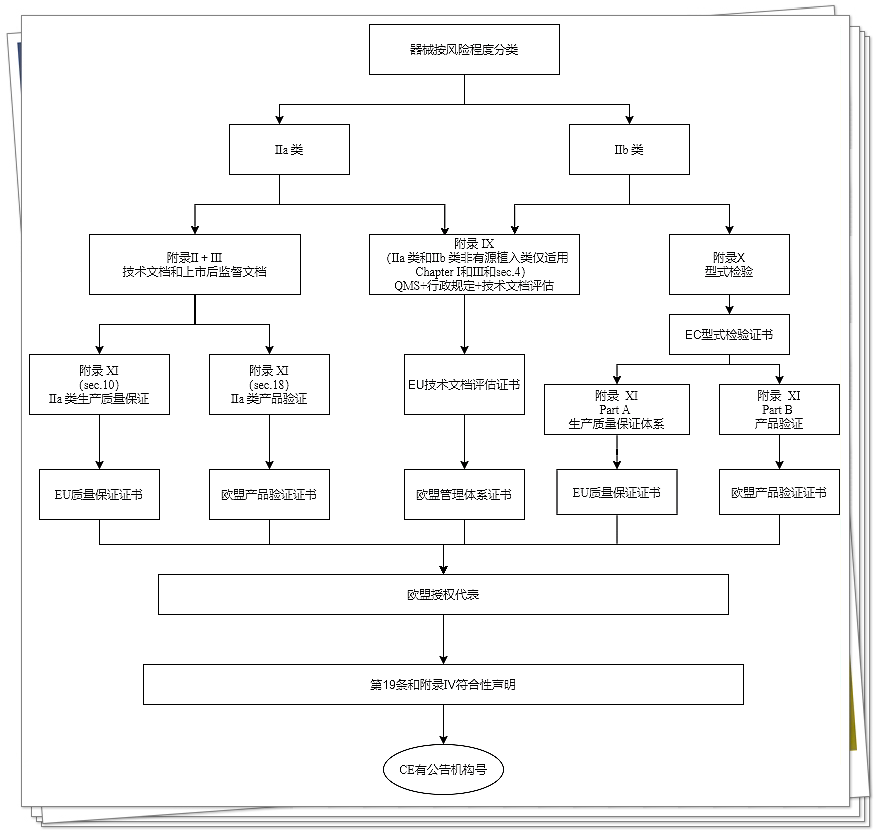
IIb类器械
IIb类器械(非客户定制类或研究类器械)的制造商,应依据附录9的第1章和第2章的规定进行符合性评估(附录第4节规定的“对各同类器械组中至少1个代表性器械的技术文档评估”)。
然而,IIb类可植入器械(除缝线、U形钉、牙齿填充物、牙套、齿冠、螺钉、楔子、牙板、金属丝、针、小夹和连接体外),附录9第4节中所规定的技术文档评估应适用于所有器械。
此外,也可选择按照附录X(基于型式检验的符合性评估)规定的符合性评估,联合附录XI(基于产品合规性验证的符合性评估)规定的符合性评估进行。
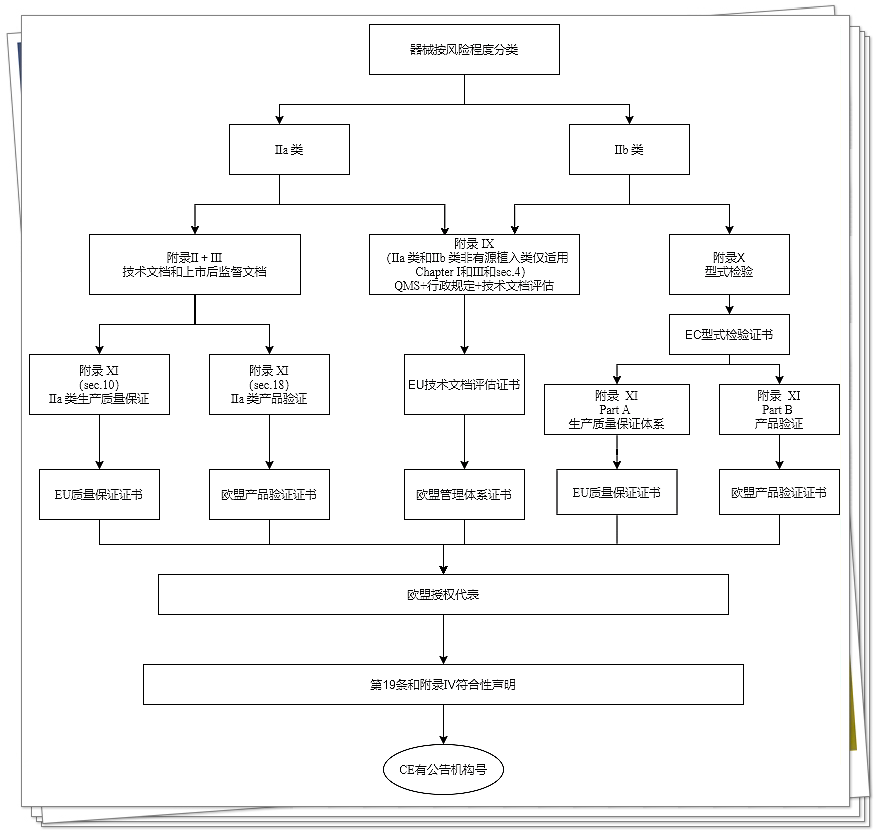
IIa类器械
IIa类器械(非客户定制类或研究类器械)的制造商应根据附录9第1和3章规定的质量管理体系进行符合性评估(附录第4节规定的“对各同类器械组中至少1个代表性器械的技术文档评估”)。
此外,制造商也可选择根据附录2(技术文档)和3(上市后监督的技术文档)及基于附录11第10节或第18节器械符合性验证的符合性评估起草技术文档。该技术文档评估应至少适用各器械类别中至少1个代表性器械。
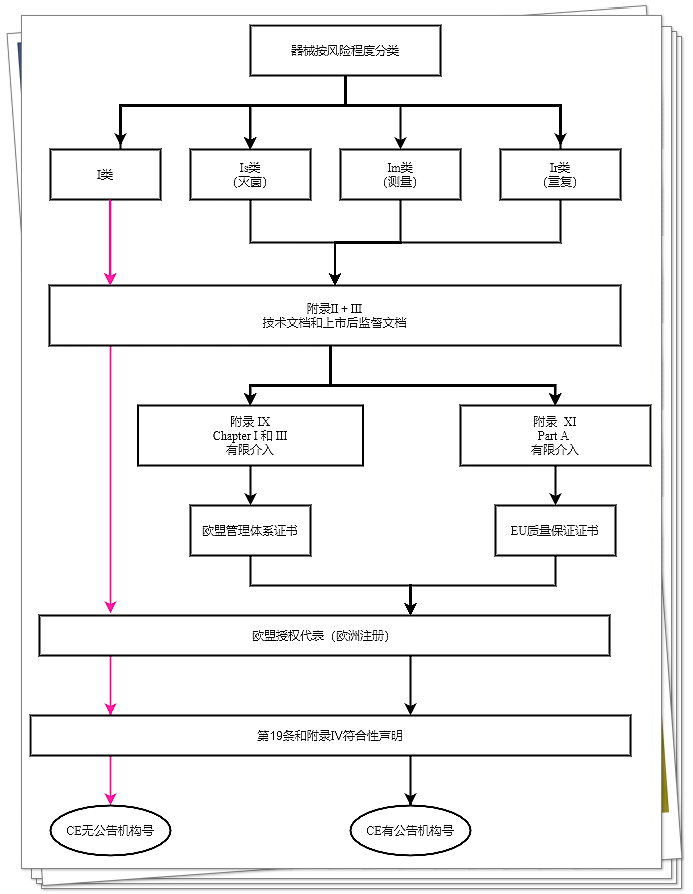
I类器械
I类器械(非客户定制类或研究类器械)的制造商在制定附录2和附录3规定的技术文档后,需通过签发第19条中的《符合性声明》,以声明其器械的符合性。
如果这些器械以无菌状态进入市场、具有测量功能或可重复使用,制造商应实行附录9第1章和第3章或附录11的A部分所述程序。
但是,公告机构的介入范围应限于:
·如果此类器械以无菌状态进入市场,则涉及无菌条件建立、保障和维持;
·如果此类器械具有测量功能,则涉及器械符合计量要求的情形;
·如果器械可重复使用,则涉及清洗、消毒、杀菌、维护、功能测试的验证及相关使用说明书。
定制器械
客户定制器械的制造商,应遵循附录13所述程序并按照该附录第1节规定起草声明后,再将该器械投放市场。
根据第1子段的适用程序,III类客户定制植入器械的制造商应遵循附录9第1章规定的符合性评估流程。
此外,根据附录11的A部分规定,制造商可选择应用符合性评估。
→ 欧盟CE证书办理快·准·好的秘诀是什么?
[久顺企管集团]是您合规路上的加速引擎!始创于1996年,近30年全球合规技术专家,近20年资深欧代,荷兰\英国\美国\中国均设公司。
■ 呈献全程高效的欧盟合规服务:
√ 欧盟CE注册取证;
√ 技术文档编写;
√ 合规策略;
√ 体系辅导;
√ 上市后监督咨询等。
■ 已成功布局欧盟临床试验渠道,提供欧盟临床试验一站式CRO服务:
√ 临床方案设计、临床试验方案的撰写;
√ 与当地实验室/医院合作,安排客户试验产品合规开展临床试验;
√ 包括但不限于:收集\整理\分析试验原始数据并出具临床试验报告。



 CHINA
CHINAUK
NL
首页|久顺集团|医疗器械|验厂咨询|欧盟代表|自由销售证书|药品|联系我们
服务热线:400-658-3933 、130-618-28828 E-mail : isosh@isosh.com 公众号:久顺集团技术服务 QQ群:
版权所有:久顺企管集团 地址:上海市东方路800号宝安大厦15层,25层 备案号:沪ICP备16003407号-1 沪公网安备 31011502005499
沪公网安备 31011502005499