


MDR简介及申请流程
CE为法文CONFORMITE EUROPEENNE的首字母缩写,表示“欧洲统一”。
CE适用区域
欧盟EU+欧洲自由贸易联盟会员国,英国脱欧后,共有31个国家。
很多除欧盟外的国家,除美国FDA、日本PAL、澳大利亚TGA等,绝大数通行欧洲颁发的自由销售证书CFS。
| 法国 | 德国 | 英国(脱欧) | 爱尔兰 | 意大利 | 比利时 | 荷兰 | 西班牙 |
| 葡萄牙 | 卢森堡 | 瑞典 | 芬兰 | 奥地利 | 波兰 | 匈牙利 | 希腊 |
| 捷克 | 斯洛伐克 | 斯洛文尼亚 | 拉脱维亚 | 立陶宛 | 塞浦路斯 | 马耳他 | 罗马尼亚 |
| 丹麦 | 爱沙尼亚 | 保加利亚 | 冰岛 | 挪威 | 瑞士 | 土耳其 | 克罗地亚 |
CE标志是一种安全认证标志,凡贴有CE标志的产品均可在欧盟各成员国内销售,无须符合各个成员国的要求。使用CE标志,实现了商品在欧盟成员国范围内的自由流通,因此CE标志被视为制造商打开并进入欧洲市场的通行证。
在欧盟市场“CE”标志属强制性认证标志,不论是欧盟内部企业生产的产品,还是其他国家生产的产品,要想在欧盟市场上自由流通,就必须加贴“CE”标志,以表明产品符合欧盟《技术协调与标准化新方法》法规的基本要求, 加贴“CE”标志必须识别很多协调标准,这是欧盟法律对产品提出的一种强制性要求。
欧盟 医疗器械法规
| 名称 | 法规 | 发布日期 | 强制实施日期 |
| 医疗器械法规 | 2017/745,MDR | 2017-4-5 | 自2021-5-26起 |
| 体外诊断器械法规 | 2017/746,IVDR | 2017-4-5 | 自2022-5-26起 |
欧盟 医疗器械协调标准
| 名称 | 协调标准 | 名称 | 协调标准 |
| 质量管理体系 | EN ISO 13485 | 临床调查 | EN ISO 14155-X |
| 包装 | EN 868-X | 风险分析 | EN ISO 14971 |
| 生物学评估 | EN ISO 10993-X | 标签&符号 | EN 1041 & ISO 15223 |
| 灭菌 | ISO 11135,ISO 11137 | 医用电气安全 | EN 60601-1 |
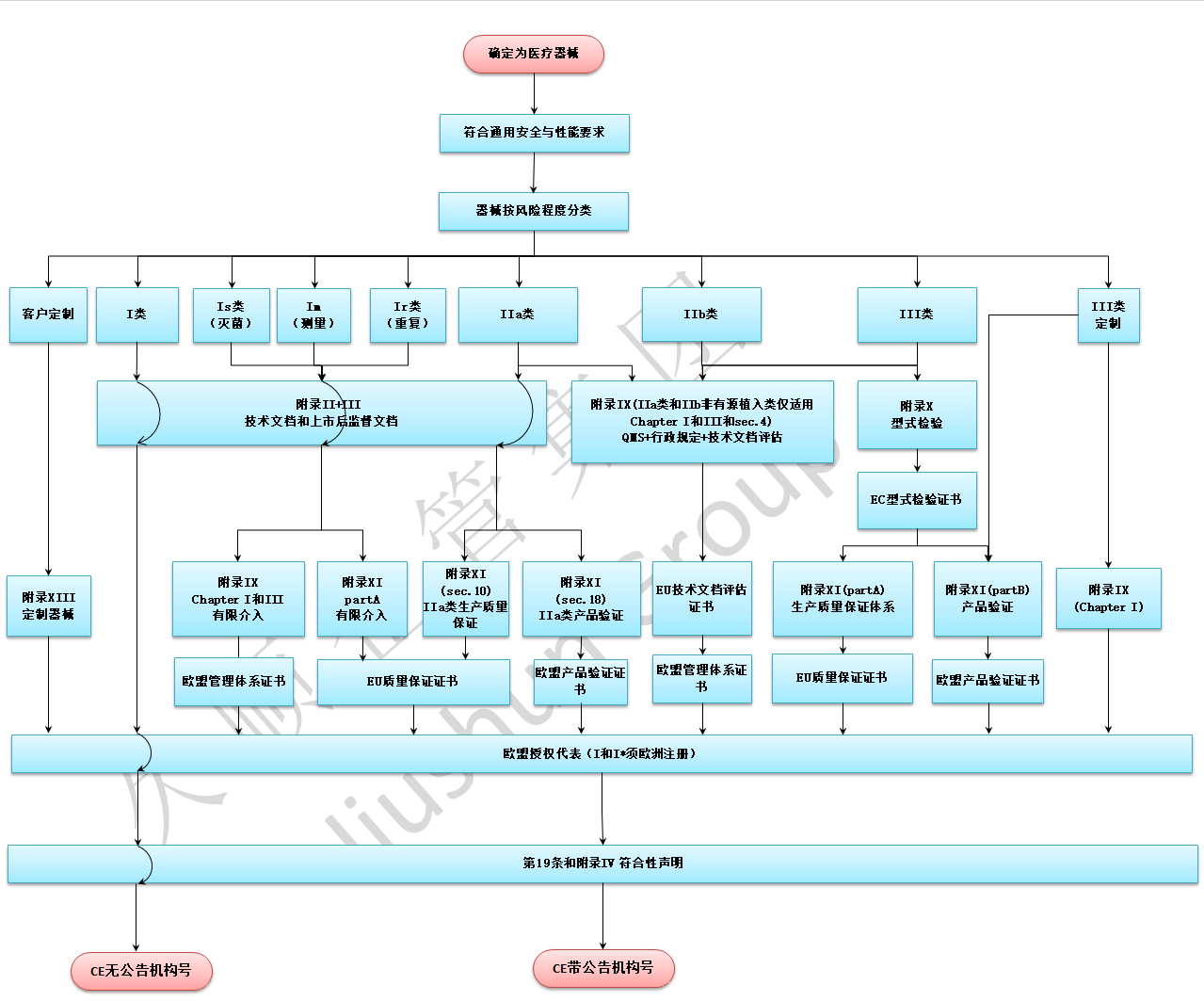
MDR管辖范围内的医疗器械,按其风险大小可以分为四个等级:I类、IIa类、IIb类、III类,其中I类的风险最低,III类最高。 这即是医疗器械的CE分类。
MDR医疗器械进行CE认证时的符合性评估分别由MDR的第 V 章规定。
普通医疗器械CE认证的一般步骤:
步骤1. 分析该器械的特点,确定它所属的法规
步骤2. 确定该器械的分类(风险分级)
步骤3. 选择相应的符合性评价程序
步骤4. 选择公告机构
步骤5. 确认适用的基本要求/有关的协调标准
步骤6. 确认该器械满足基本要求/协调标准, 并使证据文件化
步骤7. 欧盟授权代表。
步骤8. 欧洲注册。
步骤9. 对于需要公告机构评审的器械,通过公告机构的符合性程序
步骤10. 起草符合性声明并加贴CE标志
CE 技术文档要求
技术文档是欧盟医疗器械法规中很重要的一个事项,它的目的是要求企业准备充份的技术资料和证明,供主管机关调阅,或客户需要时出具。各欧盟指令对于"技术文档"的要求有所差别,在这里仅以中国出口企业最常用的“医疗器械”的要求为例,加以说明。
医疗器械法规2017/745要求“技术文档”可能包含:企业简介及欧洲代表名称、联系方式;CE符合性声明(或称自我保证声明,若该产品是和其它设备联合运用,则应有整体符合基本要求的证明材料),主要内容如下:
1.器械说明与性能指标:
包括变型和附件包含器械说明与性能指标,以及引用的前代和类似器械的信息。
2.制造商提供的信息:
产品资料和控制文档(包括产品生产工艺流程图)。
产品的灭菌方法和确认的描述。
灭菌验证。
产品质量控制措施。
产品稳定性和有效期的描述。
3.设计与制造信息:
产品的历史沿革。
技术性能参数。
产品配合使用的附件、配合件和其它设备清单。
产品的图示与样品。
产品所用原材料及供应商。
包装材料说明。
包装验证。
标签。
使用说明书。
4.通用安全与性能要求:
产品检验报告及相关文献,包含其符合附录I提供的通用安全与性能要求的证明资料。
5.风险利益分析和风险管理:
产品潜在风险测试报告及相关文献。
6.产品验证与确认:
产品临床试验报告,包括:
临床前评估:包含计划/执行/评估/报告/更新,临床前文献检索,实验室测试/模拟使用测试/计算机模拟/动物试验/与风险管理程序的接口,可用的临床前数据分析评价。
临床评估:包含临床评估计划/文献检索方法/文献检索出的文献/临床研究/验证/等效性/适用性/临床评估报告,上市后监管/PMCF计划/报告/与风险管理程序的接口,可用的临床数据分析评价。临床使用概述及权威观点。临床评估报告。
以及针对含药器械、人体/动物来源组织或其衍生物制备的器械、引入人体并被吸收器械、具有测量功能器械等的相关附加信息。
附1.产品出厂检测报告。
附2.产品型式检测报告。
附3.基本要求检查表。
7.上市后监管计划。
8.上市后监管报告或定期安全性更新报告(PSUR)。
9.符合性声明文件。
10.CE符合性标志。
11.器械的可追溯性信息(UDI)。
12.欧盟授权代表。
13.欧洲注册。
备注:
◇ 生物相容性测试(A)第一部分要求:细胞毒性、致敏、刺激、皮内反应、全身毒性(急性毒性)、亚慢性毒性(亚急性毒性)、遗传毒性、植入、血液相容性; (B)支持测试:慢性毒性、致癌性、生殖与发育毒素、生物降解。)
◇ 包装合格证明。
◇ 标签、使用说明书。
◇ 结论(技术文档的接受、利益对应风险的陈述)。
MDR分类
MDR 2017/745号法规附录VIII中详定22条规则,按医疗产品的危险程度,将产品分为Ⅰ类、Ⅱa类、Ⅱb类、Ⅲ类。
一、 产品分类规则:
1. 使用持续时间
1.1.“短暂”是指预期正常连续使用不超过60分钟。
1.2.“短期”是指预期正常连续使用60分钟到30天之间。
1.3.“长期”是指预期正常连续使用超过30天。
2. 侵入性器械和有源器械
2.1. “身体孔口”是指身体的任何天然开口,以及眼球的外表面,或者任何永久性人工开口,如造口。
2.2. “外科侵入性器械”是指
(a) 侵入性器械从身体表面穿透进身体,包括外科手术时通过身体孔口的粘膜穿透;
(b) 一种不通过身体孔口穿透的器械
2.3. “可重复使用的外科器械”是指通过切割、钻、锯、刮、削、夹、收缩、剪切或类似方式用于外科使用的器械,不连接到任何有源医疗器械,制造商预期可通过适当的处理之后再次使用,如实施清洁、消毒和灭菌。
2.4. “有源治疗器械”是指任何有源器械,无论是单独使用或与其他器械联合使用,以支持、更改、替换或恢复生物学功能或结构,以期疾病、损伤或残障得到治疗或缓解。
2.5. “用于诊断和监测的有源器械”是指任何有源器械,无论是单独使用或与其他器械组合使用,用于为检测、诊断、监测或治疗生理病症、健康状况、疾病或先天畸形。
2.6. “中央循环系统”是指以下血管:肺动脉、升主动脉、弓主动脉、动脉分岔的降主动脉、冠状动脉、颈总动脉、颈外动脉、颈内动脉、脑动脉、头臂干、心静脉、肺静脉、上腔静脉、下腔静脉。
2.7. “中枢神经系统”是指脑、脑膜和脊髓。
2.8. “损伤的皮肤或粘膜”是指皮肤或粘膜呈现病理变化或带来疾病或伤口变化的区域。
二、分类准则:
1.规则应用由器械的预期用途来决定;
2.如果器械是和其它器械配合使用,分类规则分别适用于每种器械;
3.附件可以和其它一起使用的器械分开单独分类;
4.启动或影响某种器械的软件与器械属于同一类型。
规则1~4、所有非创伤性器械均属于I类,除非他们:
用于储存体液(血袋例外)=> II a类
于Ila类或更高类型的有源医疗器械类=> II b类
改变体液成分=> II a/II b/III类
对于接触皮肤伤皮肤或粘膜的=> II a/II b类
规则5、侵入人体孔径的医疗器械
暂时使用(牙科压缩材料、检查手套)=> I类
短期使用(导管、隐形眼镜)=>IIa类,但咽部以上的口腔、耳鼓以外的耳道或鼻腔时=>I类
长期使用(正常牙线)=> II b类,但用于咽部以上的口腔、耳鼓以外的耳道或鼻腔且不易通过粘膜吸收时=>IIa类。
规则6~7、外科创伤性器械
再使用的外科器械(钳子,斧子)=> I类
暂时或短期使用(缝合针、外科手套) => 11a类
用于电离辐射形式供应能量,施用药物时 => IIb类
与中央循环系统(CCS)或中枢神经系统接触的器械 III类。
规则8、所有植入式器械和长期外科侵入性器械均归类为 IIb 类,除非其:
放置在牙齿上 => IIa类。
用于心脏或中央循环系统或中枢神经系统,在体内产生化学变化,具有生物效应或能够被完全吸收或大部分被吸收,用于施用医疗产品时,为有源植入式器械或其相关附件,为乳房植入物或心脏修补 网状织物,为完整或部分关节置换物,为直接与脊柱接触的椎间盘置换植入物或为植入器械=> III类。
规则9、用于注入或交换能量的所有有源治疗器械均归类 IIa 类,用于控制、监测或直接影响有源植入器械的性能的 => III类。
规则10、用于诊断和监测的有源器械均归类 => IIa 类,用于释放电离辐射和预期用于诊断或治疗放射的有源器械 =>IIb 类。
规则11、软件的分类进行了细化=> I/IIa/IIb/III
规则12、控制药物或其他物质进出人体的有源器械 => II a类
如以一种潜在危险方式工作 =>II b类
规则13、所有其他有源医疗器械属于=>I类
规则14、与医用物质结合的器械(含杀精子的避孕套、含抗生素的牙髓材料)=> III类
规则15、所有用于避孕或预防性病传播的器械均归类为 =>IIb 类,除非其为植入式或长期侵入性 器械,在此情形下,应归类为=> III 类。
规则16、专门用于隐形眼镜的消毒、清洗、漂洗、或水合的器械,用于侵入性器械消毒的消毒溶液或清洗消毒器=>II b类,用于医疗器械消毒或灭菌的器械均 =>IIa 类。
规则17、专门用于记录 X 射线辐射生成的诊断图像的器械均归类为 IIa 类。
规则18、 所有利用非活性或处理为非活性的人体或动物源组织或细胞或其他衍生物制成的器 械均归类为 III,除非此类器械仅用于直接接触无损皮肤。
规则19、所有纳入或包含纳米材料的器械,根据所带来的潜在的风险=>IIa/IIb/III类。
规则20、通过自然腔道进入,以呼吸方式给药的侵入器械=>IIa/IIb类。
规则21、预期通过自然腔道引入人体或应用于皮肤,并被人体吸收或 局部分散在人体内的物质或组合=>IIa/IIb/III类。
规则22、集成了诊断功能的有源治疗器械,该诊断功能对于病人管理具有重大影响的=>III类。
MDR申请流程

 CHINA
CHINAUK
NL
首页|久顺集团|医疗器械|验厂咨询|欧盟代表|自由销售证书|药品|联系我们
服务热线:400-658-3933 、130-618-28828 E-mail : isosh@isosh.com 公众号:久顺集团技术服务 QQ群:
版权所有:久顺企管集团 地址:上海市东方路800号宝安大厦15层,25层 备案号:沪ICP备16003407号-1 沪公网安备 31011502005499
沪公网安备 31011502005499