

医疗器械

体外诊断试剂注册
体外诊断试剂注册
一、注意事项:
一)、二类向申请人所在省、自治区、直辖市药监局递交注册资料,三类向国家局药监局递交注册资料;
二)、用于注册检验的器械的生产应符合医疗器械质量管理体系;
三)、需提供合格的检验报告;
四)、产品是否需要临床试验;
五)、申请人名称、注册地址应与企业营业执照一致;
六)、申报资料项目应齐全,并符合注册资料形式要求;
七)、应重视注册体系考核;
八)、应在医疗器械注册证有效期届满6个月前向原食品药品监督管理部门申请延续注册;
九)、变更向原注册部门提交。
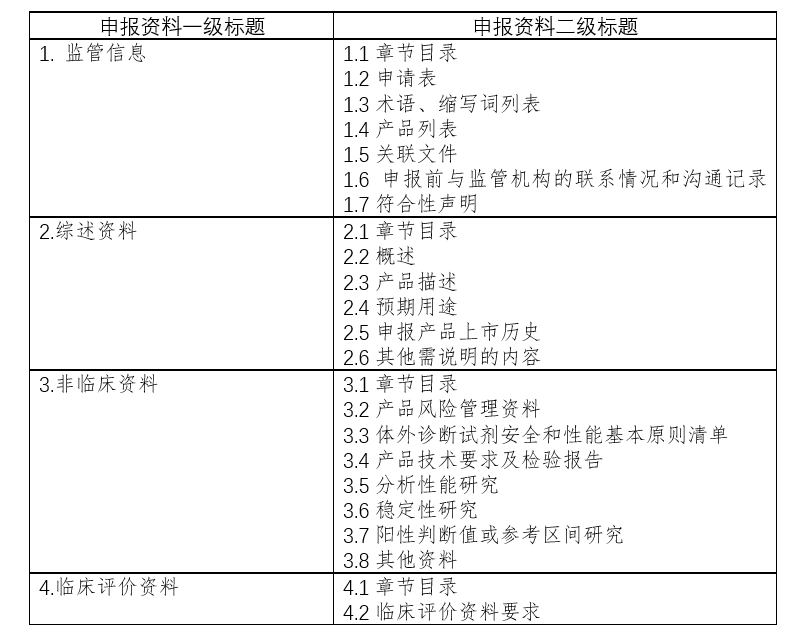
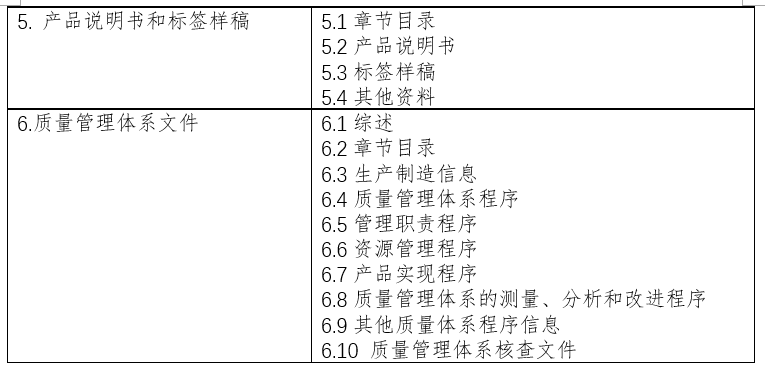
二、注册申报清单:
三、久顺企管集团技术支持:
一)、辅导办理体外诊断试剂产品注册证和生产许可证;
二)、提供体外诊断试剂注册申请资料范本;
三)、主导编写相关注册申请资料;
四)、审核申报资料;现场辅导,并向相应药监局递交资料;
五)、取得产品注册证,生产许可证。

 CHINA
CHINAUK
NL
首页|久顺集团|医疗器械|验厂咨询|欧盟代表|自由销售证书|药品|联系我们
服务热线:400-658-3933 、130-618-28828 E-mail : isosh@isosh.com 公众号:久顺集团技术服务 QQ群:
版权所有:久顺企管集团 地址:上海市东方路800号宝安大厦15层,25层 备案号:沪ICP备16003407号-1 沪公网安备 31011502005499
沪公网安备 31011502005499