


1.各类产品的延期时间
对比IVDD指令,IVDR法规存在巨大差异。其中,市场上约80%体外诊断产品的合规途径由“自我符合性声明”变为“需要公告机构审查”。
然而,目前具备IVDR认证审核资质的公告机构仅12家,公告机构审核能力的短缺使得市场需求无法得到满足。
IVDR延期草案旨在某些条件下给予制造商和公告机构更多的时间用于完成必要的合格评定程序,从而降低IVD短缺的风险,其中缓解D类器械的短缺最为紧迫。
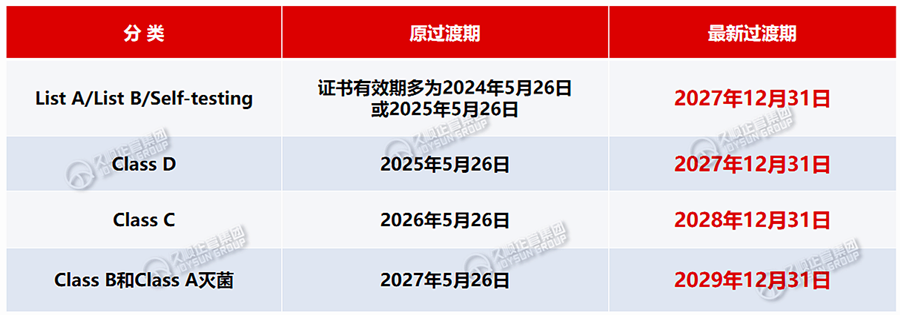
2.各类产品的延期措施
2.1 Class A灭菌、Class B、Class C、Class D
享受草案所规定新过渡期需满足的条件有:
□ 产品应继续符合98/79/EC指令;
□ 产品的设计和预期目的无重大变化;
□ 产品不会对患者、使用者或其他人的健康或安全或对保护公众健康的其他方面造成不可接受的风险;
□ 不迟于2025年5月26日,制造商已建立质量管理体系;
□ 制造商已向公告机构提交正式申请,要求对产品或旨在替代产品进行符合性评估,但不迟于以下时间:
·List A/List B/Self-testing产品、Class D产品:2025年5月26日;
·Class C产品:2026年5月26日;
·Class B及Class A灭菌产品:2027年5月26日。
□ 公告机构和制造商在以下时间内签署书面协议:
·Class D产品:2025年9月26日;
·Class C产品:2026年9月26日;
·Class B及Class A灭菌产品:2027年9月26日。
□ 制造商应建立符合IVDR法规要求的上市后监督和警戒系统。
2.2 List A/List B/Self-testing产品
□ 公告机构根据98/79/EC指令颁发的证书,在2022年5月26日后仍然有效,之后未被撤销,在证书所示期限结束后继续有效至2027年12月31日。
□ 公告机构根据98/79/EC指令颁发的证书,在2022年5月26日仍然有效,但在本草案生效日之前已过期,需符合以下2项条件中的1项,可视为有效期延长至2027年12月31日:
·在证书到期日之前,制造商和公告机构已根据IVDR法规签署书面协议,对到期证书所涵盖的器械或旨在替代该器械的器械进行合格评定。
·成员国主管当局已根据IVDR法规第54条第(1)款批准符合性评估流程的豁免,或已根据IVDR法规第92条第(2)段要求制造商执行符合性评估流程。
3.不适用延期的重大变更
产品在过渡期内发生的设计或预期用途变更,不属于重大变更的,被允许继续遵循IVDD指南在市场销售流通。
那么问题来了:哪些设计或预期用途的变更,不属于重大变更?MDCG 2022-6给出了解答!
该指南的变更讨论范围是设计或预期用途的变更。
3.1 预期用途变更
□ 不属于重大变更的预期用途变更:
预期用途的限制,例:缩小适用人群范围,临床样本类型、采样生理部位的变更。
□ 属于重大变更的预期用途变更:
·新增检测标志物;
·新增功能,例:新增检测、测量或诊断功能等;
·伴随诊断试剂扩大适用人群范围;
·新增临床样本类型;
·从定量检测变为定性检测;
·从专业人群适用变为非专业人群适用;
·从自动检测变为手动检测。
3.2 设计用途变更
□ 设计变更分为:设计变更、软件变更、成分或原材料变更、灭菌方式变更。
□ 不属于重大变更的设计变更:
未改变产品操作原理,即未对安全性能造成新的不良影响、或未对产品性能中风险与获益判定造成负面影响,例如:
·孵育温度和时间的变更;
·新增清洗步骤等类似的操作步骤变更;
·为更高效控制温度,对PCR新循环周期的变更;
·从冷藏变为室温保存;
·通过上市后监督收集到的数据,在说明书上对交叉干扰物质进行更精准描述的变更;
·对试剂盒组成成分数量的变更。
【注意】以上变更仅为列举,并不包含所有现实可能遇到的情况。
□ 属于重大变更的设计变更:
·从手动操作方式变为半自动或全自动操作;
·反应条件的变化(例:时间、温度等);
·通过上市后监督收集到的数据,在说明书上对产品灵敏度进行下调处理的变更;
·由于产品特异性降低,定性产品cut-off值的变更。
4.IVDR的质量体系升级要求
■ 不迟于2025年5月26日,制造商已建立质量管理体系(EN ISO 13485+IVDR使用条款)。
■ 针对质量管理体系,IVDR法规第10条第8点明确要求:制造商应确保采取必要流程,使产品系列的生产符合本法规要求。
■ 制造商的质量管理相关人员应当充分知晓质量体系流程和程序,重点工作有:
·首先,根据EN ISO13485:2016适用要求建立QMS;
·其次,审查适用的MDR\IVDR法规条款,并确认QMS程序和记录的欠缺之处。



 CHINA
CHINAUK
NL
首页|久顺集团|医疗器械|验厂咨询|欧盟代表|自由销售证书|药品|联系我们
服务热线:400-658-3933 、130-618-28828 E-mail : isosh@isosh.com 公众号:久顺集团技术服务 QQ群:
版权所有:久顺企管集团 地址:上海市东方路800号宝安大厦15层,25层 备案号:沪ICP备16003407号-1 沪公网安备 31011502005499
沪公网安备 31011502005499