


欧盟发布MDCG 2025-5文件《IVDR下体外诊断医疗器械性能研究的问答》,以下是对该文件的核心要点提炼。

一、性能研究是什么?
1. 定义
- 性能研究(Performance Study):旨在确立IVD器械的分析或临床性能的研究(IVDR第2(42)条)。
- 分析性能研究:验证器械检测/测量特定分析物的能力(如灵敏度、特异性、精密度等)。
- 临床性能研究:验证器械结果与特定临床状况或生理或病理过程或状态相关的能力(如诊断灵敏度、阳性预测值等)。
2. 适用范围
- 适用于所有符合"性能研究"定义的活动,对发起人身份无限制(如学术界、企业)。
- 不适用场景有:
·早期设计验证阶段(未涉及性能确认)。
·纯实验室工作流程研究(非器械性能评估)。
·符合IVDR第5(5)条的"内部制造"IVD(需符合通用安全和性能要求GSPR且仅限医疗机构使用)。
二、对性能研究有哪些关键的监管要求?
1. 需何时提交申请(Application)或通报(Notification)?
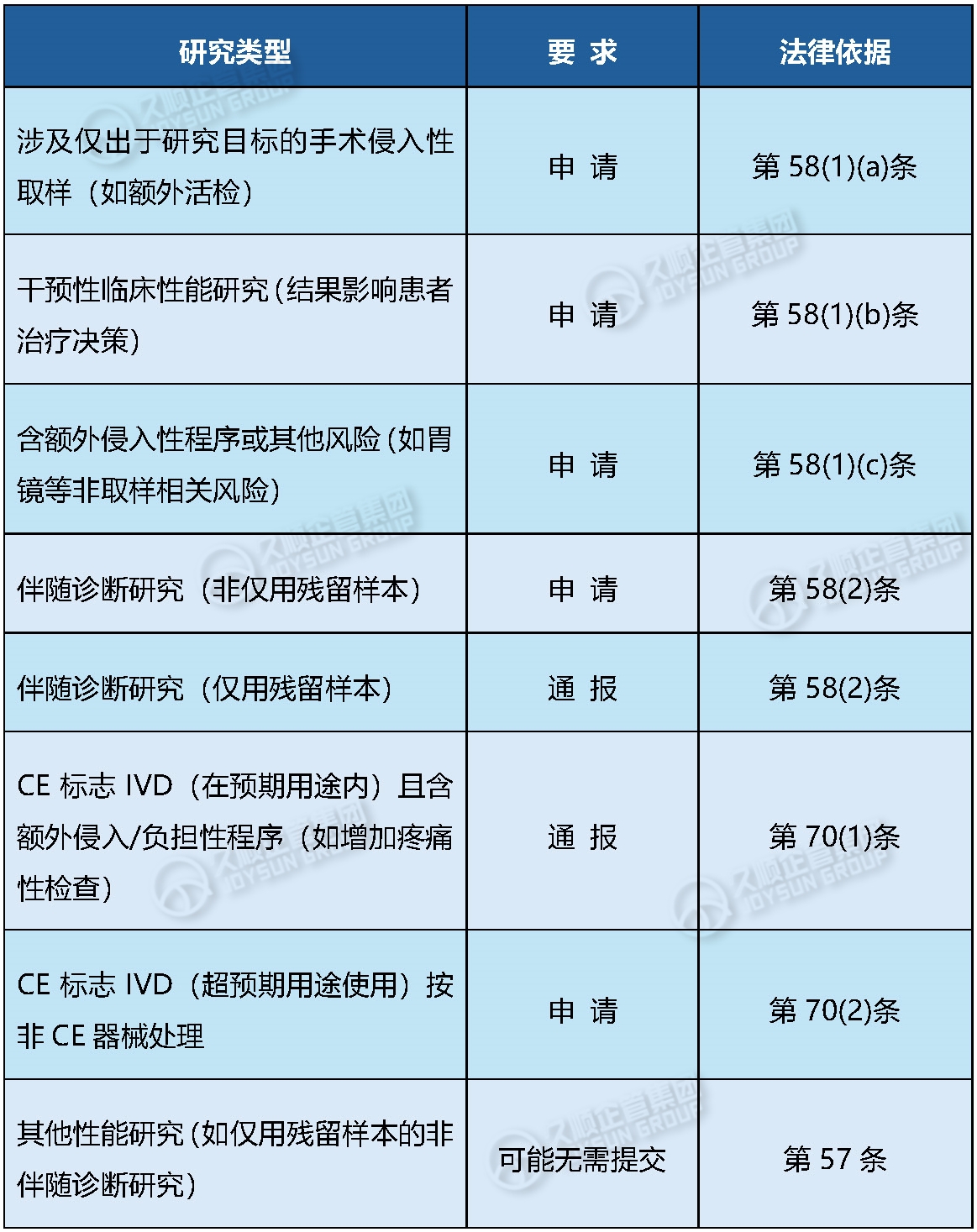
流程图参考:附录I的决策树(Page 31)作为核心判定工具。
2. 特殊概念定义
- 残留样本(Left-over Samples):临床常规剩余样本(无个体临床需求),在非干预性研究中使用。
- 伴随诊断:起到安全有效使用对应药物的功能的IVD(如识别药物受益患者)。
- 负担性程序(Burdensome):引起疼痛、不适、干扰生活的程序(需从受试者角度评估)。
3. 多成员国研究要求
- 样本采集国:需提交申请/通报(因涉及受试者)。
- 仅样本分析国:通常无需提交(除非涉及伴随诊断残留样本研究)。
三、性能研究操作流程有哪些重要事项?
1. 启动时间
- 研究启动日=首个成员国招募起始日(或首例残留样本分析日)。
- 需确保分析中心已就绪后,方可开始样本采集。
2. 文件要求
- 申请材料:需包含附件XIII第2-3节及附件XIV文件(如研究计划、研究者手册)。
- 性能评估计划(PEP):无需提交,但需备查。
- 标签与说明书:需根据成员国要求翻译(即使仅采集样本)。
3. 安全与伦理
- 安全性报告:遵循MDCG 2024-4指南。
- 伦理审查:IVDR未作统一规定,需遵守各国要求。
四、性能研究涉及哪些变更管理?
1. 实质性修改(Substantial Modification)
- 定义:可能影响受试者安全/权利或数据可靠性的变更(如变更主要终点、器械设计)。
- 流程:
·需在文件更新后1周内通报成员国。
·38天后未收到异议即可实施(含7天专家咨询期)。
- 例外情况:仅用残留样本的伴随诊断研究无需通报变更。
2. 非实质性修改:无统一要求,需遵循各国规定。
注意:附录II列出可能构成实质性修改的变更清单(如终点调整、受试者人数变化等)。
五、性能研究的过渡与特殊场景是什么?
- 遗留研究:2022年5月26日前已批准的IVDD研究无需重新提交,但需按IVDR报告严重不良事件。
- RUO(仅限研究使用)产品转化:
·若产品被赋予IVD预期用途,则视为IVD并需遵守IVDR。
·三种使用场景:纯研究(非IVDR)、转为研究对象(需合规)、转为研究工具(需CE或内部制造)。
- 联合研究:如果联合研究在一个地点进行,申办者可以为性能研究和临床试验/临床研究指定不同或相同的研究者。被指定的调查员需具备相应法规所规定的必要资格。共同文件(如知情同意书)的修改需分别评估是否触发CTR/IVDR变更流程。
>>注意:
1. 以上要求需结合成员国具体法规(尤其是伦理审查、非实质性修改)。
2. 分析性能研究同样受IVDR约束(如符合第58(1)标准则需申请)。
3. EUDAMED生效前使用CIV-ID或国家ID作为研究识别号。
IVDR性能研究·久顺出品,合规✔可信✔
√久顺是国内少有的配备IVDR法规\IVD技术\数据分析领域专业团队的企业,已与欧盟各大主流实验室和医院建立合作渠道,可提供临床性能研究(国内少有可胜任且使用EP文件研究)、可用性研究、上市后临床跟踪研究等高效服务。
→案例是最有力的代言↘
√久顺已成功辅导优思达获TÜV南德签发的IVDR CE证书(涵盖10项产品),并助力其取得国内核酸检测领域首张IVDR荷兰CIBG注册证书。久顺为优思达提供了精湛的13485体系+IVDR辅导(CE技术文档辅导)、欧代等关键性合规服务。
√久顺已成功辅导江苏宏微特斯获得BSI签发的HCG早早孕自测试剂盒B类IVDR CE证书,通过差异性分析攻克欧盟临床难题,为客户顺利完成ISO13485结合IVDR法规适用条款的升级,并配合企业成功通过现场审核,仅用时9个月超前通过IVDR CE审核。


 CHINA
CHINAUK
NL
首页|久顺集团|医疗器械|验厂咨询|欧盟代表|自由销售证书|药品|联系我们
服务热线:400-658-3933 、130-618-28828 E-mail : isosh@isosh.com 公众号:久顺集团技术服务 QQ群:
版权所有:久顺企管集团 地址:上海市东方路800号宝安大厦15层,25层 备案号:沪ICP备16003407号-1 沪公网安备 31011502005499
沪公网安备 31011502005499