


2025年12月,欧盟医疗器械协调小组(MDCG)正式发布 MDCG 2025-10 指南文件,对医疗器械(MDR)与体外诊断医疗器械(IVDR)的上市后监督(PMS) 进行系统、全面的阐述。该文件并非新增法规要求,而是对 MDR/IVDR 中 PMS 相关条款的权威解读与实操指引。

一、为什么PMS如此重要?
1.首先明确PMS在欧盟法规体系中的核心地位:
•PMS 是 质量管理体系(QMS)不可分割的一部分
•覆盖器械整个生命周期
•目的是确保器械在真实世界使用中的安全性、性能与获益-风险比持续可接受
2.法规明确要求制造商:
•主动、系统性 收集上市后数据
•利用数据持续更新:风险管理、临床/性能评价、技术文档
•必要时采取 CAPA 或 FSCA
3.PMS不再是“被动处理投诉”,而是一个持续运行的闭环系统。
二、适用范围与目标
1.适用范围:
•适用于所有医疗器械(MD)和体外诊断器械(IVD)
•包括所有风险等级
•涵盖定制器械
2.指南目标:
·解释什么是 PMS 系统
·说明PMS 计划的构成
·描述PMS 的主要活动
·阐明PMS 与 QMS 其他模块的 联动关系
3.明确不涵盖:
•PSUR / PMS Report 的具体写作模板(已有其他 MDCG 指南)
•医疗机构自制器械(Article 5(5))
三、法规要求下的PMS系统
1. PMS 的基本要求
制造商必须:
•规划、建立、实施、维护并持续更新 PMS 系统
•PMS 的深度与复杂度必须 与器械风险等级和类型相匹配
PMS 系统应当能够:
•主动收集真实世界数据
•分析质量、性能与安全性
•支持是否需要采取行动的决策
2. PMS 与 QMS 的关系
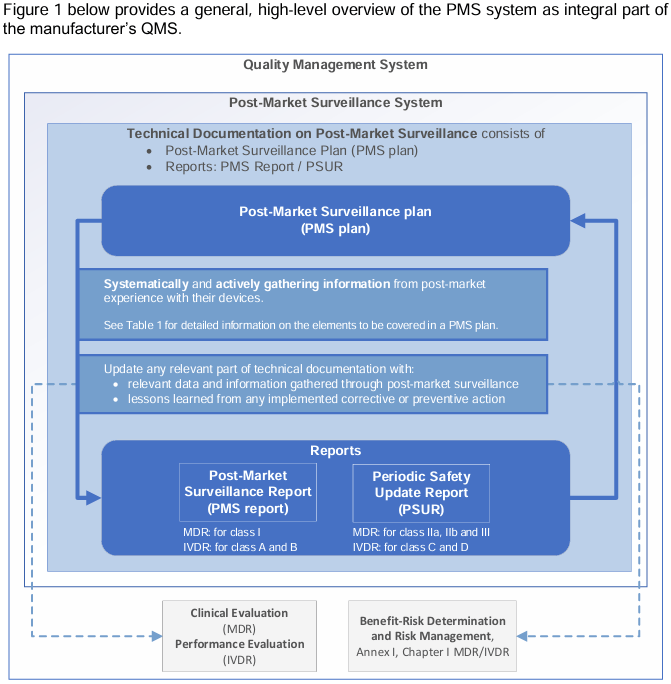
指南强调:
•PMS 是 QMS 的输入源
•PMS 数据必须反馈至:风险管理、临床 / 性能评价、设计变更、标签和说明书、CAPA 与 FSCA
未深度融合QMS的PMS将被视为不合规。
四、PMS 计划
1.PMS 计划是:
•PMS 系统的基础文件
•技术文档的一部分(MDR Annex III / IVDR Annex III)
2.PMS 计划应回答的关键问题:
•监测哪些内容?
•使用哪些数据来源?
•采用什么分析方法?
•频率如何?
•达到什么阈值需要行动?
•谁负责?如何记录?
3.指南强调的三个关键词:
•Proactive(主动)
•Systematic(系统性)
•Risk-based(基于风险)
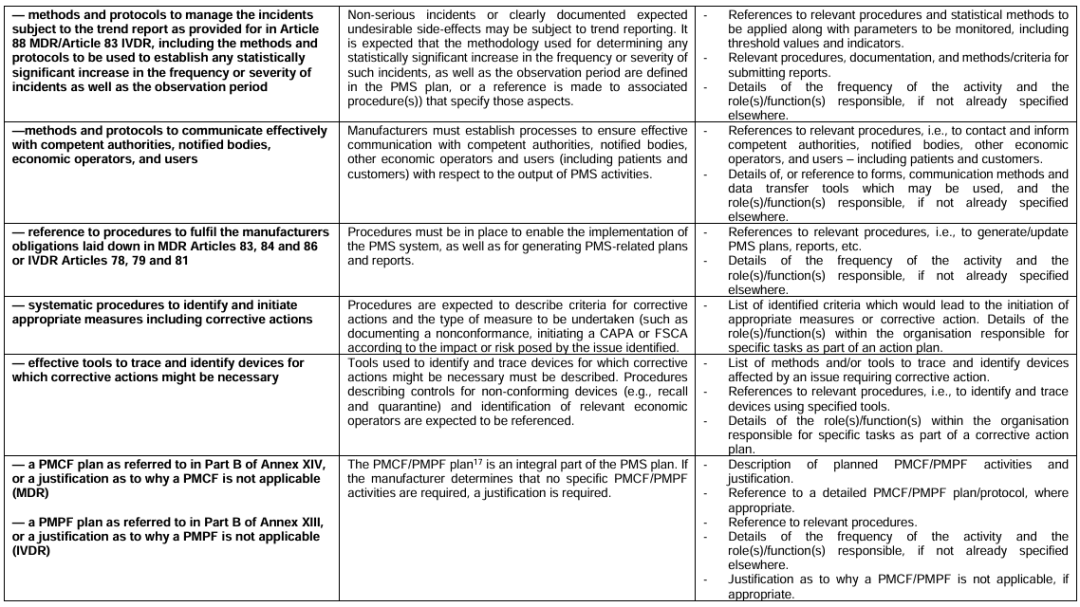
4.PMS 计划必须覆盖的要素(高度概括):
•数据来源(投诉/文献/注册数据库/用户反馈等)
•数据分析方法(定性/定量)
•指标与阈值(趋势/风险变化)
•投诉与事件调查方法
•趋势报告方法
•沟通机制(CA/NB/用户)
•CAPA / FSCA 触发机制
•PMCF / PMPF 计划或合理性说明
PMS Plan ≠ 程序文件!而是 “做什么 + 为什么这样做” 的总纲文件。

五、PMS 的主要活动
指南将 PMS 运行拆解为 4大连续步骤:
1. 确定数据来源
可能包括:
•严重事件与 FSCA
•非严重事件与投诉
•文献与数据库(如 EUDAMED)
•类似产品的公开信息
•用户、经销商、进口商反馈
特别提醒:谨慎使用社交媒体等非可验证数据来源
2. 数据收集
•PMS 数据通常从器械首次上市后开始
•对高风险或创新器械:需要通过 PMCF / PMPF 主动生成数据
3. 数据分析与评估
分析目标包括:
•性能是否持续符合预期?
•是否出现新的风险或副作用?
•获益-风险比是否仍然可接受?
•是否落后于“技术现状(SOTA)”?
4. 结论与行动
基于分析结果,制造商必须判断:
•是否需要更新风险管理?
•是否启动 CAPA / FSCA?
•是否需要修改设计、IFU、标签?
•是否需要更新 PMS 计划本身?
所有结论必须体现在:
•PMS Report 或 PSUR 中
六、PMS 与 QMS 其他模块的联动
PMS 数据必须用于:
•更新风险管理与获益-风险评估
•更新设计、制造信息
•更新临床/性能评价
•更新 SSCP(如适用)
•识别改进器械可用性与性能的机会
•识别对其他器械的潜在影响
•识别趋势并履行趋势报告义务
七、附录与参考文件
Annex 1
•MDR / IVDR 下 PMS、PMS Plan、PMS Report、PSUR 的义务对照表
适合作为内部培训或合规检查清单
Annex 2
•通过 IVD 与植入器械的示例场景
•展示 PMS 如何触发:风险更新、召回、通知公告、设计变更
总 结
从 MDCG 2025-10 可以清晰看出三大趋势:
1. PMS 更强调“主动性”
2. PMS 必须深度融入QMS
3. 真实世界数据成为合规核心证据
PMS 不再是“上市后的合规负担”,而是贯穿产品生命周期的核心管理工具。
警戒系统\上市后监督\经济运营商\器械登记等,都是CE所要求质量体系的重要组成。如需体系建立\培训\升级等服务,可咨询[久顺企管集团]。
久顺已为诸多制造商完成MDR/IVDR体系升级、公告机构体系监督审核中的MDR/IVDR发补意见,已建立完备的MDR\IVDR体系升级服务:GAP分析表、上市后监督系统、警戒系统等。
具体服务包括:上市后监督计划PMS Plan; 上市后性能/临床跟踪计划PMPF\PMCF Plan; 趋势报告Trends Report; 定期总结报告Periodic Summary Report等记录报告建立\执行\培训。
久顺是您合规路上的加速引擎!始创于1996年,30年全球合规技术专家,近20年资深欧代,西班牙\荷兰\英国\美国\中国均设公司。全程高效的欧盟合规服务:欧盟CE注册取证、技术文档编写、合规策略、体系辅导、上市后监督咨询等。


 CHINA
CHINAUK
NL
首页|久顺集团|医疗器械|验厂咨询|欧盟代表|自由销售证书|药品|联系我们
服务热线:400-658-3933 、130-618-28828 E-mail : isosh@isosh.com 公众号:久顺集团技术服务 QQ群:
版权所有:久顺企管集团 地址:上海市东方路800号宝安大厦15层,25层 备案号:沪ICP备16003407号-1 沪公网安备 31011502005499
沪公网安备 31011502005499