


欧盟IVDR联合实施和准备计划-
第二期 指导文件和行动
B组:
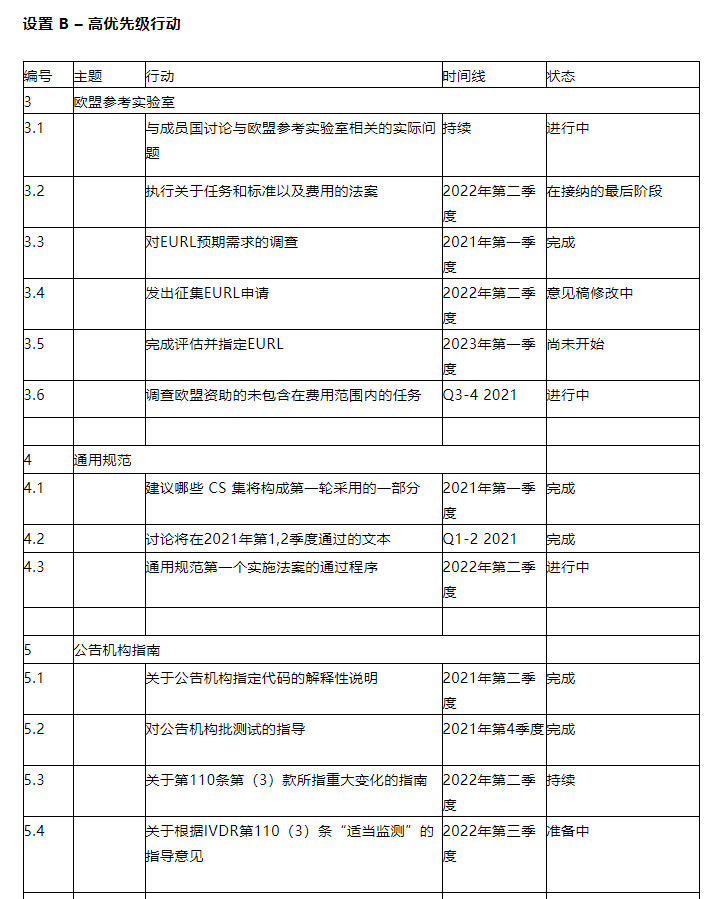
欧盟参考实验室
IVDR 规定,委员会可以指定一种新型的独立科学机构,即欧盟参考实验室。
欧盟参考实验室以前从未在体外诊断领域建立过。
这些实验室指定后,将对属于其指定范围的D类器械进行额外检测,尤其是将D 类器械投放市场之前验证的性能以及是否符合任何适用的通用规范。
此外,他们将对带有CE标志的 D 类器械投放市场之前的样品或批次进行测试。
欧盟参考实验室还将提供一系列专业咨询功能。因此,在欧盟,它们的建立对于D类器械进行高水平、连贯性的评估非常重要。
通用规范
通用规范是对合格评定某些要素的具有法律约束力的要求,以实施法案的形式采用。
公告机构和欧盟参考实验室将根据通用规范评估器械,并且它们的存在使涵盖的器械免于专家小组咨询的额外步骤。
指令 98/79/EC(第 2002/364/EC 号指令)下存在一系列的通用技术规范。
此外,MDCG 的IVD子组正在根据该组认可的路线图开发D类器械的新通用规范。
目前尚无草案可用的其他 CS 将在以后的轮次中开发和采用。
公告机构指南
针对 MDR和 IVDR 通用的方面大量指导已经发布或正在MDR联合实施计划下准备。
本节考虑针对 IVD 行业的公告机构的重要指南。已经制定了器械分类指南。
另一个重要的指导要素涉及制造商、公告机构和新成立的欧盟参考实验室之间在测试方面的互动,它旨在阐明 IVDR 中规定的每个参与者的责任以及互动的实用性。
此外,关于第 110 条第 3 款第 2 项,公告机构、制造商以及指定机构迫切需要明确“对其已认证器械相关的所有适用要求进行适当监督”的含义。在这种情况下,指导文件应详细说明指定机构将开展的活动,作为适当监督的一部分,并涵盖有关某些制造商义务的要求,特别是关于其质量管理体系的要求。
性能评估及专家小组
IVDR加强了对器械临床证据的要求。
例如,它规定了性能评估的三个要素:科学有效性、分析性能和临床性能,并就如何证明这些要素提出了详细要求。
需要包括性能评估计划、性能评估报告和上市后性能跟踪计划在内的文件。
D类器械的性能将由欧盟参考实验室进行验证。
对于非常新颖的高风险器械,作为合格评定的附加要素,公告机构必须就制造商的性能评估报告咨询专家小组。
IVDR 第 29 条规定了对 C 类和 D 类器械的新文件的要求——安全和性能摘要。
它将由指定机构进行评估,并在EUDAMED上公开发布。
由于这是一项新要求,因此应制定有关如何构建安全和性能摘要的指南。这应该建立在MDCG CIE WG 中对医疗器械的安全性和临床性能进行同样的总结的工作的基础上。
IVDR 首次规定,当 EUDAMED 此类功能运行时,通过 EUDAMED 就某些性能研究进行申请或通知,此规定是欧盟级别的要求。
标准
提供欧盟官方公报 (OJEU) 中引用的协调标准(这些标准可以佐证符合性假设),这将给造商符合 IVDR 的要求提供帮助。为此,委员会必须要求相关的欧洲标准化组织(CEN 和 Cenelec)修订现有标准并制定新标准,随后在 OJEU 中公布 MDR 和 IVDR 下的协调标准参考清单,以持续定期更新和扩大。
伴随诊断
伴随诊断器械在IVDR中被定义为安全和有效使用相应医药产品所必需的器械,用于在治疗之前和/或治疗期间识别最有可能从该药品中受益的患者或可能在治疗中受益的患者。
使用相应的药物治疗会增加严重不良反应的风险。
虽然它们只占体外诊断市场的一小部分,但它们对于正确使用相应的药品以及让患者获得定制化的、更有效的治疗非常重要。它们的发展与药品的发展有着内在的联系。
鉴于对此类产品性能评估的强化要求以及要求公告机构需就伴随诊断器械的适用性,咨询药品权威机构或欧洲药品管理局 (EMA) 的新要求,需要对伴随诊断提供指南。
作为首要任务,为了让这些器械完成合格评定,应制定一个基本的咨询流程。
遗留器械
IVDR 第 110(3) 条规定,对于 2022 年 5 月 26 日之前投放市场的器械以及根据过渡性规定可以合法投放市场的器械(遗留器械),“本法规有关上市后监督、市场监督、警戒、经济经营者和器械注册的要求应适用于本段第二和第三小段所指的器械,而不是指令 98/79/EC 中的相应要求”。
EUDAMED
EUDAMED在 2022年5月26日之前无法全面运行,并且有第113 (3)(f) 条 IVDR 的过渡性规定,因此该系统的使用将在第 34(3)条MDR中提及的通知发布后六个月内才会成为强制性或可执行的。
因此,在EUDAMED完全发挥作用之前,就统一的管理实践和信息交换的替代技术解决方案提供指导将是有益的。该指南应使成员国和其他相关方能够有效地履行其在IVDR下的义务,同时最大限度地减少相关方的任何潜在额外负担。
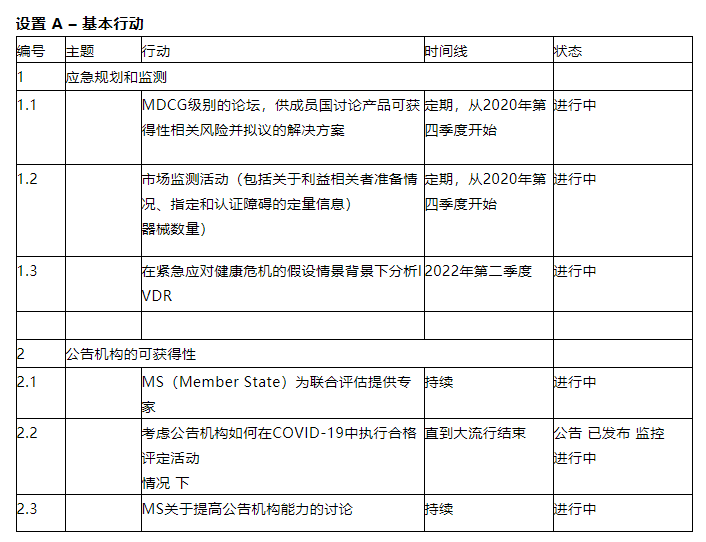
后附摘要简表,可以对计划实施情况做快速的了解。


结语
结束语:
我们拥有专业的技术团队,编写技术文件由久顺企管集团IVD技术部完成,企业可以提供中文/英文资料,我们提供重要文件英文编写服务,技术文件(含DOC)输出结果为英文;完成产品注册由久顺企管集团旗下荷兰子公司Lotus NL B.V.完成,Lotus提供欧代服务和注册服务,产品注册最终取得CIBG注册证书。
离2022年5月19日取证,剩下1个月的时间,时间非常紧迫,请抓紧时间办理!
制造商要在2022年5月19日前取证,应该快速行动,把需要办证的产品罗列清楚,久顺企管集团国内外技术团队为您提供欧代服务、技术文件编写和产品注册一站式快捷合规服务。办证周期:1-2个月内完成(不含节假日)。
IVDD Other CE注册收益:可节省大量费用,可使用周期长:
5月19日之前在欧盟相关当局注册,按照在IVDR法规下的分类分段适用IVDR法规:
(a) D类产品有效期到2025年5月26日;
(b) C类产品有效期到2026年5月26日(严重的精神药物检测试剂盒);
(c) B类产品有效期到2027年5月26日(适用于药物滥用检测试剂盒);
(d) A类灭菌产品有效期到2027年5月26日。

 CHINA
CHINAUK
NL
首页|久顺集团|医疗器械|验厂咨询|欧盟代表|自由销售证书|药品|联系我们
服务热线:400-658-3933 、130-618-28828 E-mail : isosh@isosh.com 公众号:久顺集团技术服务 QQ群:
版权所有:久顺企管集团 地址:上海市东方路800号宝安大厦15层,25层 备案号:沪ICP备16003407号-1 沪公网安备 31011502005499
沪公网安备 31011502005499