


附件
医疗器械注册自检工作规定
(征求意见稿)
为加强医疗器械(含体外诊断试剂)注册管理,规范注册申请人注册自检工作,确保医疗器械注册审查工作有序开展,根据《医疗器械监督管理条例》《医疗器械注册管理办法》《体外诊断试剂注册管理办法》,制定本规定。
一、自检能力要求
(一)总体要求
对于注册时开展自检的,注册申请人应当具备自检能力,并将自检工作质量管理纳入医疗器械生产质量管理体系,配备与产品检验要求相适应的检验设备和设施,具有相应质量检验部门或者专职检验人员,严格检验过程控制,确保检验结果真实、准确、完整和可追溯,并对自检报告负主体责任。
(二)检验能力要求
1.人员要求。注册申请人应当具备与所开展检验活动相适应的管理人员和检验人员。注册申请人应当配备专职检验人员,检验人员应当为正式聘用人员,并且只能在本企业从业。
检验人员的教育背景、技术能力和数量应当与产品检验工作相匹配。检验人员应当熟悉医疗器械相关法律法规、标准和产品技术要求,掌握检验方法原理、检测操作技能、作业指导书、质量控制要求、实验室安全与防护知识、计量和数据处理知识等,并且应当经过医疗器械相关法律法规、质量管理和有关专业技术的培训和考核。
检验人员、审核人员、批准人员等应当经注册申请人依规定授权。
2.设备和环境设施要求。注册申请人应配备满足检验方法中要求的仪器设备和环境设施,建立和保存设备及环境设施的档案、操作规程、计量/校准证明、使用和维修记录,并按有关规定进行量值溯源。
开展特殊专业检验的实验室,如生物学评价、电磁兼容、生物安全性实验室等,其环境设施条件应符合其特定的专业要求。
3.样品要求。注册申请人应当建立并实施检验样品管理程序,确保样品受控并保持相应状态。
4.检验质量控制要求。注册申请人应能使用适当的方法和程序开展所有检验活动。适当时,包括测量不确定度的评定以及使用统计技术进行数据分析。
鼓励注册申请人参加由能力验证机构组织的有关检验能力验证/实验室间比对项目,提高检测能力和水平。
5.记录的控制要求。所有质量记录和原始检测记录以及有关证书/证书副本等技术记录均应归档并按适当的期限保存。记录包括但不限于设备使用记录、检验原始记录、检验用的原辅材料采购与验收记录等。记录的保存期限不应低于相关法规要求。
(三)管理体系要求
对于注册时开展自检的,注册申请人应当按照有关检验工作和申报产品自检的要求,建立和实施与开展自检工作相适应的管理体系。
自检工作管理体系应纳入医疗器械生产质量管理体系。注册申请人应当制定与自检工作相关的质量管理体系文件,包括政策、程序、作业指导书等,所开展检验工作的风险管理及医疗器械相关法规要求的文件等,并确保其有效实施和受控。
(四)自检依据标准
注册申请应依据拟申报注册产品的产品技术要求进行全项目检验。
检验方法的制定应与相应的性能指标相适应。应优先考虑采用已颁布的标准检验方法或公认的检验方法。
检验方法应当进行适当的验证或确认,确保检验具有可重复性和可操作性。
体外诊断试剂类产品,检验方法中还应明确说明采用的参考品/标准品、样本制备方法、使用的试剂批次和数量、试验次数、计算方法等。
(五)其他
1.委托生产的注册申请人可以委托受托生产企业开展自检,并出具相应自检报告。
2.境内集团公司或其子公司通过中国合格评定国家认可委员会(CNAS)认可的实验室,或者境外集团公司或其子公司具有通过境外政府或政府认可的相应实验室资质认证机构认可的实验室的,集团公司或其子公司经授权,可以由相应实验室开展自检,由注册申请人出具相应自检报告。
二、自检报告要求
(一)首次注册时提交的自检报告应当是符合产品技术要求的全项目检验报告。
变更注册、延续注册按照相关规定提交相应检验报告。报告格式应当符合检验报告模板(见附件1)的要求。
(二)自检报告应当结论准确,便于理解,用字规范,语言简练,幅面整洁,不允许涂改。签章应当符合《医疗器械注册申报资料和批准证明文件格式》《体外诊断试剂注册申报资料和批准证明文件格式》相关要求。
(三)自检报告中的检验型号、规格/包装规格应当可以覆盖注册单元内其它型号。
三、委托检验要求
(一)委托条件
注册申请人提交自检报告的,若不具备产品技术要求中部分条款的检验能力,可以将相关条款委托有资质的医疗器械检验机构进行委托检验。有资质的医疗器械检验机构应符合《医疗器械监督管理条例》第七十五条中规定。
(二)对被委托方的评价
注册申请人应当在医疗器械生产质量管理体系文件中对被委托方的资质、检验能力符合性等进行评价,并建立合格被委托方名录,保存评价记录和评价报告。
(三)样品一致性
注册申请人应当确保自行检验样品与委托检验样品一致性。应当与相关被委托方及时沟通,通报问题,协助做好有关检验工作。
注册申请人应当对被委托方出具的报告进行汇总,并形成完整的自检报告。涉及委托检验的项目,其内容可以填写为见相应委托检验报告,并提供委托检验报告原件。
四、申报资料要求
注册申请人通过自检方式提交注册检验报告的,应当提交以下申报资料:
(一)自检报告。有委托检验项目的,应当注明委托的检验机构,并提供相关检验机构的资质证明文件。
(二)具有相应自检能力的声明。注册申请人应当承诺具备产品技术要求中相应具体条款项目自行检验的能力,包括具备相应人员、设备、设施和环境等,并按照质量管理体系要求开展检验。
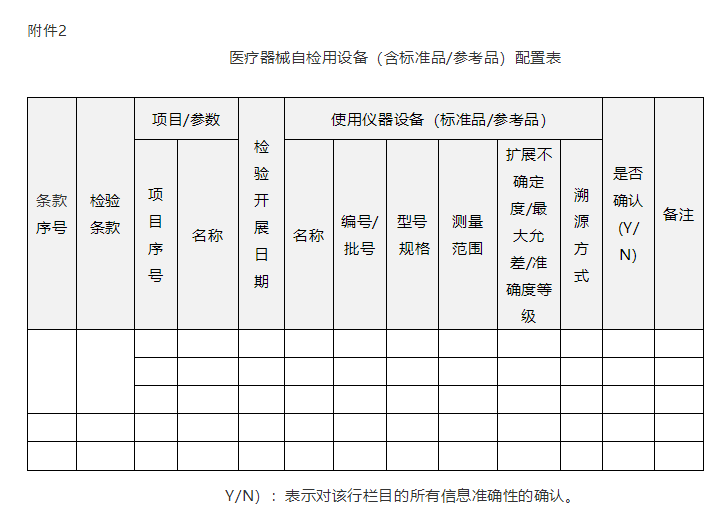
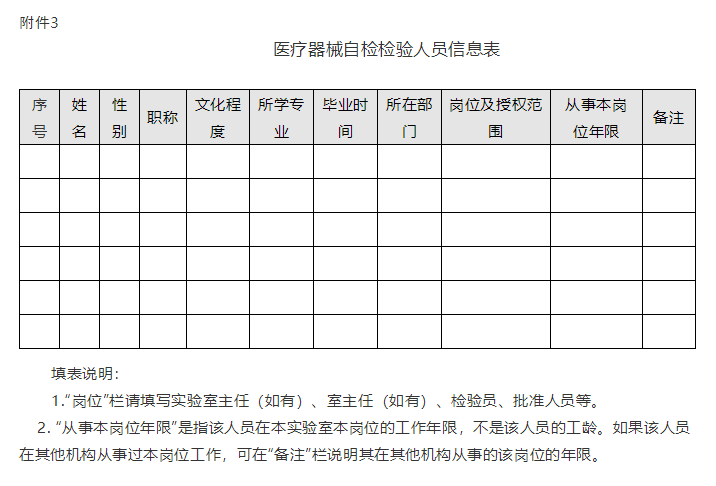
(三)质量管理体系相关资料。检验用设备(含标准品/参考品)配置表(附件2);用于医疗器械检验的软件,应当明确其名称、发布版本号、发布日期、供应商或代理商等信息(格式参考附件2);检验人员信息表(附件3);检验相关的质量管理体系文件清单,如质量手册、程序文件、作业指导书等,文件名称中应包含文件编号信息等。
(四)关于型号覆盖的说明。提供型号覆盖的相关资料,包括典型性的说明、被覆盖型号/配置与主检型号/配置的差异性分析及符合性评价等。
(五)报告真实性自我保证声明。若注册申请人将相关项目进行委托检验,自我保证声明应当包括提交自行检验样品、委托检验样品一致性的声明。
境内注册申请人自身开展自检的实验室如通过中国合格评定国家认可委员会(CNAS)认可,或者境外注册申请人自身开展自检的实验室通过境外政府或政府认可的相应实验室资质认证机构认可,可不提交本条第(二)和(三)项内容,但应当提交相应认可的证明性文件及相应承检范围的支持性资料。集团公司或其子公司经集团公司授权由相应实验室开展自检的,应提交授权书。
五、现场检查要求
对于提交自检报告的,药品监管部门开展医疗器械注册质量管理体系现场核查时,除按照有关医疗器械注册质量管理体系核查指南要求办理外,还应当对注册申请人自检能力和自检情况进行核查,重点关注检验记录、检验质量控制能力、检验人员操作技能、检验人员资质要求、设施和环境、检验仪器和设备等。
(一)检验记录:查看原始记录,检验设备使用、校准、维护和维修记录,检验环境条件记录,检验样品的有效性、对被委托方审核评价记录和报告,委托检验报告(如有),委托检验协议(如有)等。
(二)质量控制能力:查看检验相关的质量手册、程序文件、标准、作业指导书(如适用)、操作规程、检验方法验证/确认记录、内部质量控制记录等文件。
(三)检验人员操作技能:对声称自检的项目进行随机抽查,要求检验人员信息表中相应检验人员根据作业指导书(或操作规程),对留样样品或自检样品进行现场操作,应能重复检验全过程,检验方法符合要求,且检验结果与企业申报注册资料中的结论一致。
(四)检验人员资质要求:查看检验人员的在职证明、相关人员信息表中检验人员与批准人员培训记录、个人档案等文件,并与相应人员进行面对面交流,核实资质、能力是否符合有关质量管理体系要求。
(五)设施和环境:开展特殊专业检验的实验室,如生物学实验室、电磁兼容试验室、体外诊断试剂实验室等,检查实验室的设施、环境及监测记录等是否符合产品检验的要求。
(六)检验设备:核对申报资料中提交的自检用设备配置表中信息与现场有关设备是否一致。查看检验设备的检定/校准记录、计量确认资料是否满足检验要求。核查检验设备的清单,清单上应注明设备的来源(自购/长期租赁),并查看相应的合同文件。
使用企业自制校准品、质控品、样本处理试剂等的,应当查看相关操作规程、质量标准、配制和检验记录。应当关注校准品制备、量值传递规程、不确定度要求、稳定性研究等内容。关注质控品制备、赋值操作规程、靶值范围确定、稳定性研究等内容。
境内注册申请人自身开展自检的实验室如通过中国合格评定国家认可委员会认可,或者境外注册申请人自身开展自检的实验室通过境外政府或政府认可的实验室认证机构认可,按照医疗器械注册质量管理体系核查指南要求办理。
六、责任要求
注册申请人应按照《医疗器械监督管理条例》要求,加强医疗器械全生命周期质量管理,对研制、生产、检验等全过程中医疗器械的安全性、有效性依法承担责任。
自检报告虚假的,依照《医疗器械监督管理条例》第八十三条中的规定处罚。被委托方出具虚假检验报告的,依照《医疗器械监督管理条例》第九十六的规定处罚。
附件:1.医疗器械注册自检报告模板
2. 医疗器械注册自检用设备(含标准品/参考品)
配置表
3.医疗器械注册自检检验人员信息表
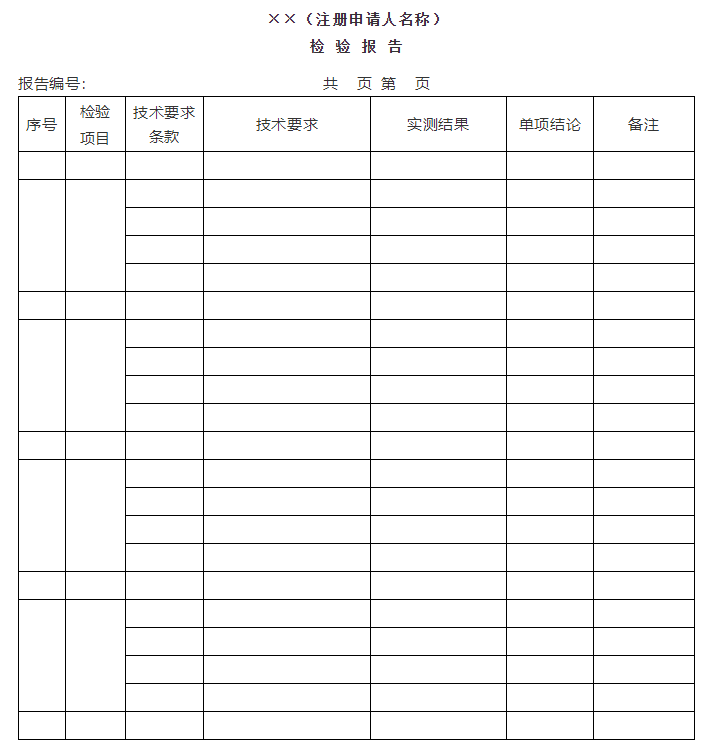
附件1
医疗器械注册自检报告(模板)
报告编号:××××
注册申请人
样品名称
型 号
生产地址
邮政编码
检验类别
声明
一、注册申请人承诺报告中检验结果的真实、准确、完整和可追溯。
二、报告签章应符合有关规定。
三、报告无批准人员签字无效。
四、报告涂改无效。
五、对委托检验的样品及信息的真实性,由委托方负责。


 CHINA
CHINAUK
NL
首页|久顺集团|医疗器械|验厂咨询|欧盟代表|自由销售证书|药品|联系我们
服务热线:400-658-3933 、130-618-28828 E-mail : isosh@isosh.com 公众号:久顺集团技术服务 QQ群:
版权所有:久顺企管集团 地址:上海市东方路800号宝安大厦15层,25层 备案号:沪ICP备16003407号-1 沪公网安备 31011502005499
沪公网安备 31011502005499